If you use PockDrug for publication, please cite:
Hiba Abi Hussein†, Alexandre Borrel†, Colette Geneix, Michel Petitjean, Leslie Regad, Anne-Claude Camproux*. PockDrug-Server: a new web server for predicting pocket druggability on holo and apo proteins. Nucl. Acids Res. May 8, 2015.
Introduction
Predicting protein pocket’s ability to bind drug-like molecules with high affinity, i.e., druggability, is of major interest in the target identification phase of drug discovery (9). Therefore, pocket druggability investigations represent a key step of compound clinical progression projects. Currently computational druggability prediction models are attached to one unique pocket estimation method despite pocket estimation uncertainties. In this site, we propose “PockDrug-Server” that predicts pocket druggability, efficient on both; estimated pockets guided by the ligand proximity (extracted by proximity to a ligand from a holo protein structure using several thresholds) and estimated pockets not guided by the ligand proximity (based on amino atoms that form the surface of potential binding cavities). PockDrug-Server provides consistent druggability results using different pocket estimation methods. It is robust with respect to pocket boundary and estimation uncertainties, thus efficient using apo pockets that are challenging to estimate. It clearly distinguishes druggable from less druggable pockets using different estimation methods and outperformed recent druggability models for apo pockets. It can be carried out from one or a set of apo/holo proteins using different pocket estimation methods proposed by our webserver or from any pocket previously estimated by the user.
You can submit your job in the druggability computation tab.
Methodology
The following flowchart describes the general workflow of PockDrug-Server:
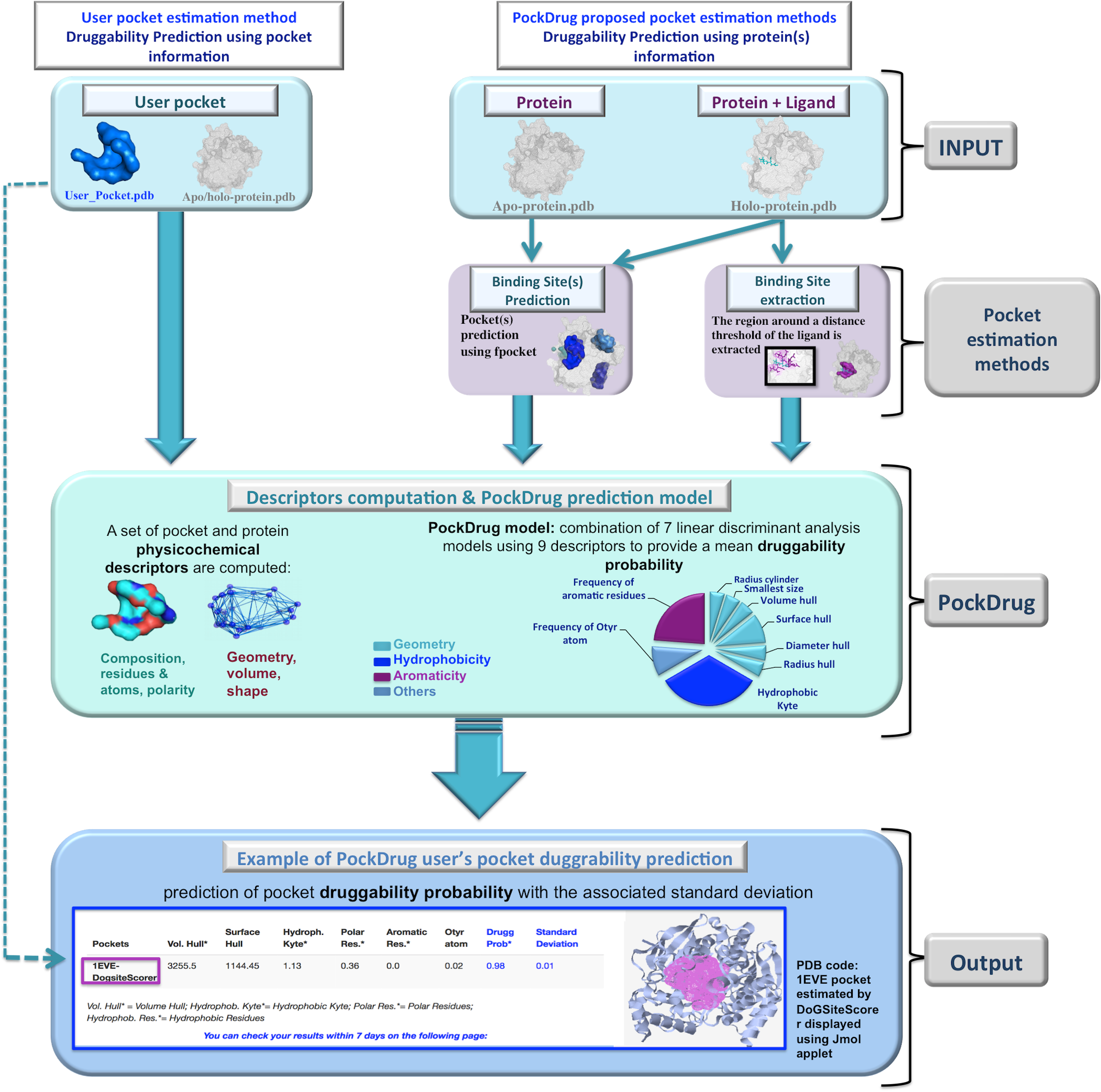
PockDrug workflow is divided into three different phases: (a) identifying protein pocket(s) (Input & pocket estimation methods) , (b) PockDrug: fast computation of physico-chemical and geometry descriptors to characterize pocket(s) (c) Output: pocket(s) druggability prediction. In the first phase two query types can be submitted:
- Druggability prediction using pocket estimated by the user
- Druggability prediction using protein structure
PockDrug: a model for predicting pocket druggability that overcomes pocket estimation uncertainties. J. Chem. Inf. Model., 10.1021/ci5006004.Borrel,A., Regad,L., Xhaard,H.G., Petitjean,M. and Camproux,A.-C. (2015)
User guide: Input, options and output
In the following section input, options and output details will be displayed in order to help the user to understand each of the steps involved and infer the output of the results produced in the workflow.
The main function of PockDrug-Server is to predict pocket druggability through the Druggability section. To do so, two types of query can be submitted:
- Pocket structure
- Protein structure
PockDrug-Sever allows users to predict druggability probability for a protein pocket estimated using an estimation pocket approach/ software of his choice. In this case, the required input is the , which corresponds to a PDB format file listing the atom pocket coordinates. The corresponding protein PDB file is also required to compute pocket descriptors and its predicted druggability probability.
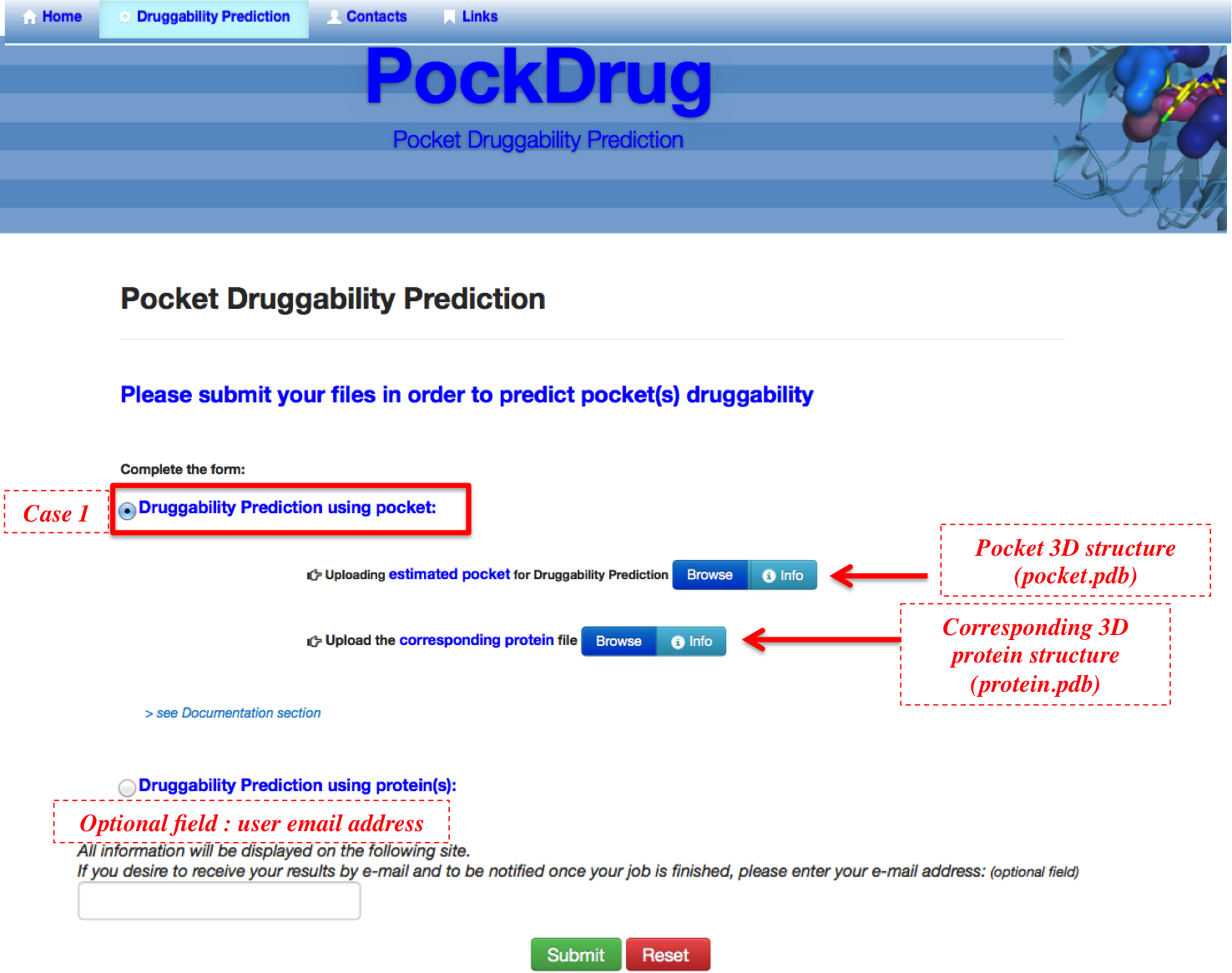
The protein structure corresponds to:
- a PDB code;
- a PDB protein file;
- or a file of PDB code list.
- Step 1: pocket(s) estimation using one or both different pocket estimation methods proposed by our web server;
- Step 2: pocket druggability probability prediction. For each pocket, previously estimated in step 1, pocket druggability probability and standard deviation are provided by PockDrug model.
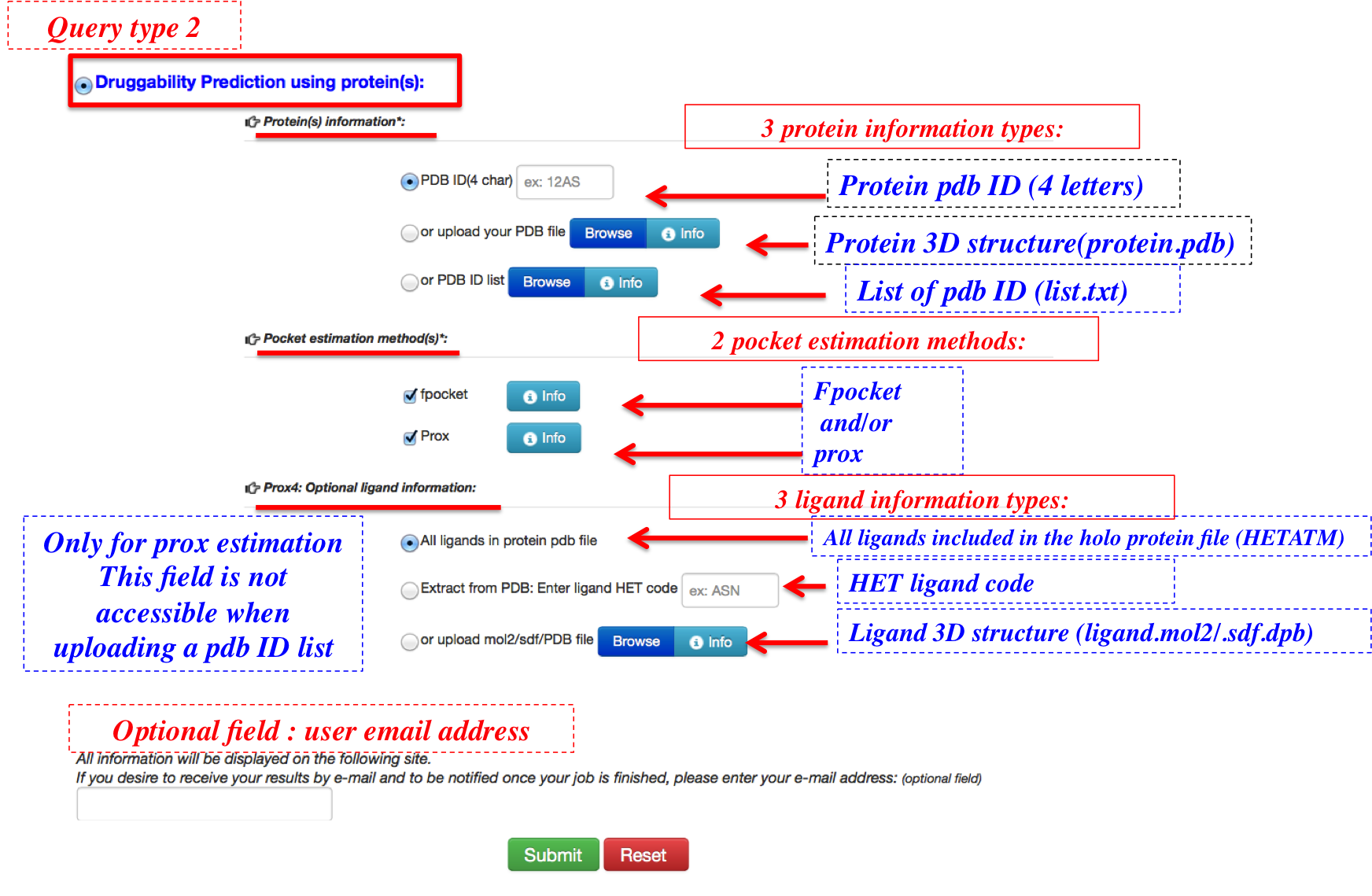
For these three types of protein structure information, prox and/or fpocket estimation methods can be choosen.
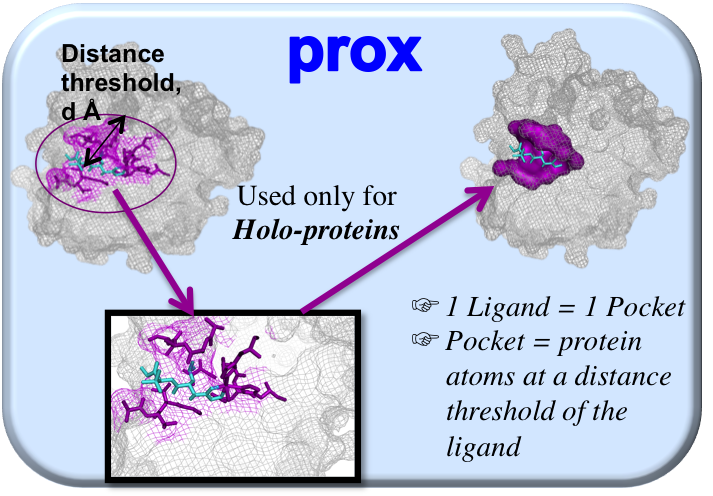
This method is based on ligand proximity information giving the user the possibility of choosing a threshold going from 4 Å to 12 Å (by step of 0.5 Å) in order to extract the protein atoms localized within the chosen distance of the ligand. Indeed, this threshold choice was recently shown to have a strong influence on the pocket descriptors (2) and it seems pertinent to give the user the opportunity to choose it. Two commonly used distance thresholds are recommended: 4 Å as used by Krasowski et al. (3), to enable the extraction of a well-defined pocket limited to short ligand interactions (as hydrogen bonds or ionic interactions) and 5.5 Å, to enable the identification of all significant contact points and a more complete environment of the binding site. This method is suitable for holo-proteins and threshold of 4 Å is chosen by default.
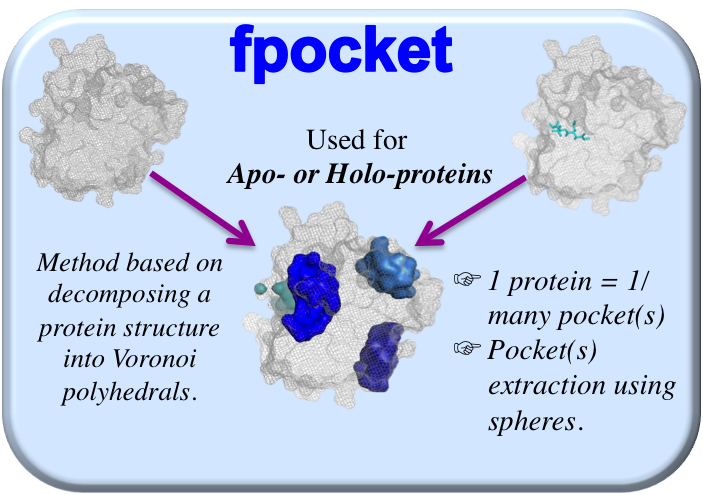
not guided by the ligand information, is an automated geometry-based method based on the decomposition of a 3D protein into Voronoi polyhedrals. It extracts all the pockets from the apo- or holo- protein surface using spheres of varying diameters. Its advantages include calculation speed and satisfactory performance in terms of overlaying known binding sites with the predicted sites (4). This method is used by default since it is suitable for both apo- and holo- proteins.
Only in single entry mode (submission of PDB ID or PDB structure), and when prox estimation method is checked, the field allowing user to submit ligand information is visible. In multiple entry mode, where a PDB ID code list is provided, and this optional ligand field is no more accessible.
3 types of ligand information input are possible:
- Default option: All ligand in the holo protein structure having "HETATM" label are considered
- Ligand HET code
- Ligand structure, in a pdb mol or sdf format
In the figure 3 and 6, user can find an amount of Help text necessary for each input field so he can easily submit his job.
After this first stage of pocket estimation, in the background some geometrical and physico-chemical pocket descriptors and protein descriptors involved in PockDrug model are computed, in the aim to predict pocket druggability. In the following part, the output details will be displayed.
The output page may consist of one or two tab(s), varying accordingly to the choice of one or two estimation method(s): one result tab per selected estimation method. Relative to the input type, two result displays are possible:
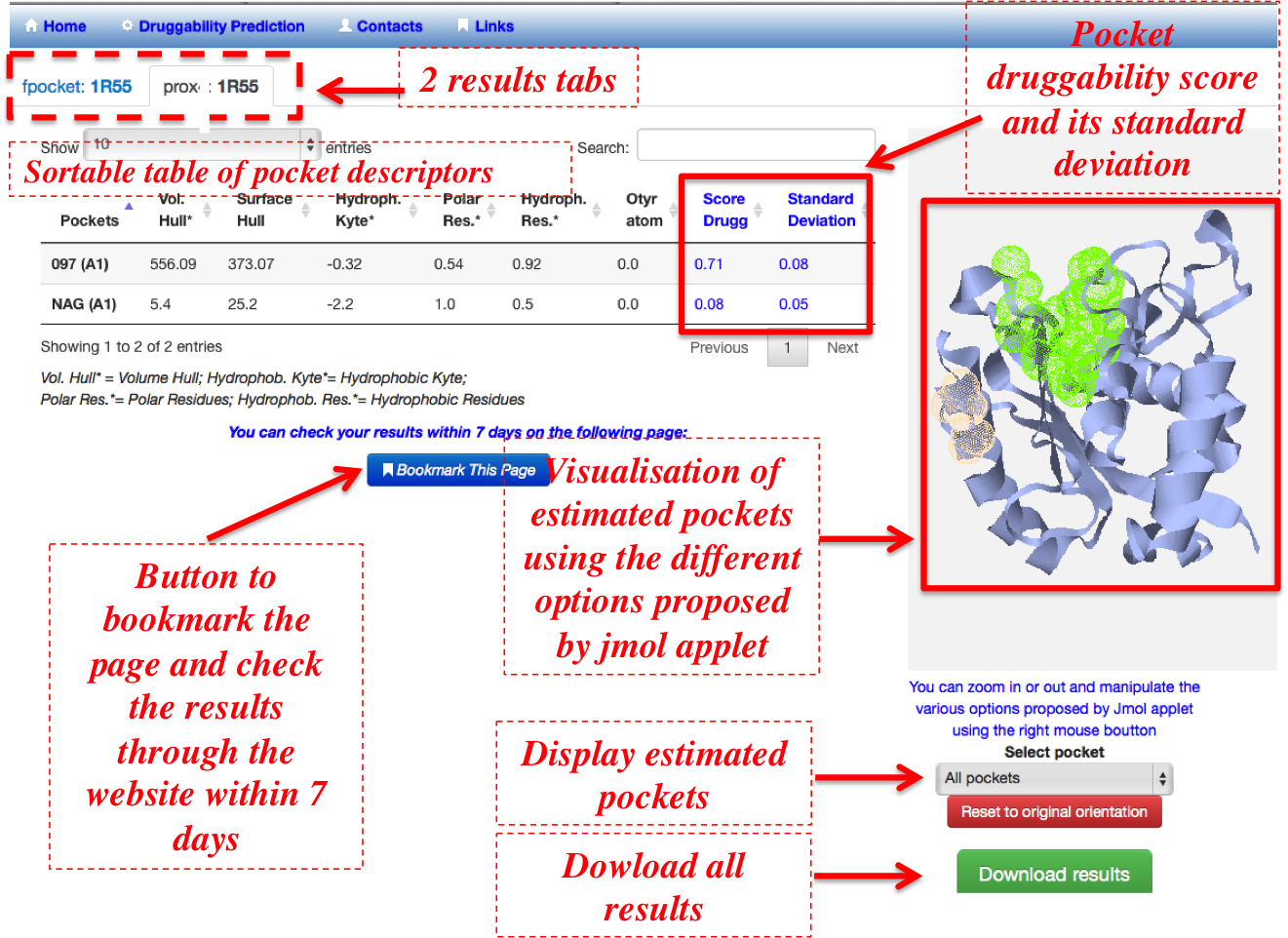
- As shown in the figure 7, each tab is structured as following:
- A sortable table providing for each protein pocket:
- Six out of the eighteen pocket descriptors involved in PockDrug model. As pocket estimation method affects directly the descriptors values, the descriptor averages with associated standard deviations computed on NRDLD set estimated using three different estimations (prox4, prox5.5 and fpocket) are given as reference (Analysis help section) to facilitate the user analysis of the pocket descriptor values.
- The average druggability probability and its associated standard deviation that indicates the druggability probability confidence on the seven best models included in PockDrug. For a probability greater than 0.5, pockets are considered as druggable. In the case where several pockets are considered, the table can be ordered in ascending or descending order of druggability probability to facilitate the identification of druggable pockets.
- Pocket visualization using the Jmol web browser applet (5) pocket(s) and protein structures can be visualized and manipulated on the server through jmol applet. All computed results: pockets structures, eighteen descriptors and druggability scores can be downloaded.
- Compressed file containing all the results can be downloaded using the download button. Only when the pockets are estimated using both fpocket and prox (for all distance threshold), overlapping scores between two pocket estimations are also computed and provided to the user through the compressed result file in order to allow pocket estimation comparisons and correspondence between two estimation methods. See section of pocket comparison in the supplementary data for the definition of overlapping scores. A bookmark button saving the link on which the user can follow the evolution of his job, access and download it within 7 days.
- Protein PDB code: giving access to the detailed result page as it is described previously in this paragraph (case a)
- Number of estimated pockets (for each method)
- Number of druggable pockets (druggability probability greater than 0.5)
- The highest druggability probability and its standard deviation
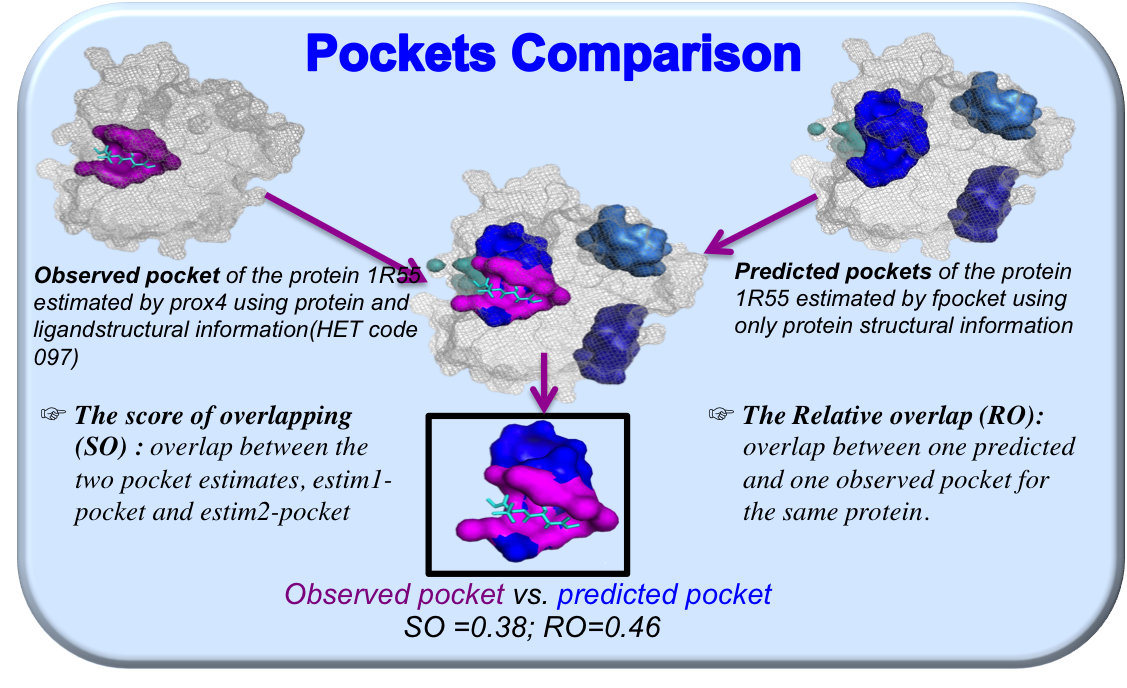
If different pocket estimation methods are applied, pockets comparison is possible through the overlapping scores. Figure 8 shows a pocket of 1R55 binding the ligand "097", estimated by prox (in magenta) and fpocket (in blue).
These overlappping scores are only included in the downloaded compressed results file.
If two default pocket estimation methods (prox and fpocket) are tested, overlapping scores between these two pocket estimations are computed and included in the downloaded compressed result file, in order to compare pocket estimates. The overlap between two estimated pockets was quantified using two scores:
- Score of Overlap (SO) indicates the overlap between the two pocket estimates, i.e., pocket1 and pocket2, as follows:
- Relative Overlap (RO) was defined by Schmidtke et al. and indicates the overlap in terms of the exposed atoms between the two estimated pockets for the same binding site:
- Mutual overlap (MO) was defined by Schmidtke et al. (6), complementary to the RO.
where Npocket1 and Npocket2 are the number of atoms in pocket1 and pocket2, respectively, and Ncommon is the number of atoms common to pocket1 and pocket2. SO yields values between 0 and 100%. An SO value of 100% indicates maximum overlap between the pair of estimated pockets used.
where SApocket1 and SApocket2 are the solvent-accessible areas of pocket1 and pocket2, respectively, computed using NACCESS software. An RO value closer to 100% indicates all exposed area in pocket1 are included in pocket2.
In the Table 1, each pocket descriptors provided by PockDrug-server is defined.
References
- Borrel,A., Regad,L., Xhaard,H.G., Petitjean,M. and Camproux,A.-C. (2015) PockDrug: a model for predicting pocket druggability that overcomes pocket estimation uncertainties. J. Chem. Inf. Model., 10.1021/ci5006004.
- Krotzky,T., Rickmeyer,T.T., Fober,T. and Klebe,G. (2014) Extraction of Protein Binding Pockets in Close Neighborhood of Bound Ligands Makes Comparisons Simple due to Inherent Shape Similarity. J. Chem. Inf. Model., 54, 3229–3237.19.
- Krasowski, A.; Muthas, D.; Sarkar, A.; Schmitt, S.; Brenk, R. DrugPred: a structure-based approach to predict protein druggability developed using an extensive nonredundant data set. Journal of chemical information and modeling 2011, 51, 2829–42.
- Le Guilloux,V., Schmidtke,P. and Tuffery,P. (2009) Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics, 10, 168.
- Herráez,A. (2006) Biomolecules in the computer: Jmol to the rescue. Biochem. Mol. Biol. Educ., 34, 255–261.
- Schmidtke,P. and Barril,X. (2010) Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J. Med. Chem., 53, 5858–67.
- Kyte,J. and Doolittle,R.F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol., 157, 105–32.
- Milletti,F. and Vulpetti,A. (2010) Predicting polypharmacology by binding site similarity: from kinases to the protein universe. J. Chem. Inf. Model., 50, 1418–31.
- Abi Hussein H, Geneix C, Petitjean M, Borrel A, Flatters D, Camproux AC. Global vision of druggability issues: applications and perspectives. Drug Discov Today. 2017 Feb;22(2):404-415.
- Hussein HA, Borrel A, Geneix C, Petitjean M, Regad L, Camproux AC. PockDrug-Server: a new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Res. 2015 Jul 1;43(W1):W436-42.